{kind=link}
{kind=link}
What are the Basic Facts about Sickle Cell Disease?
Sickle Cell Disease (SCD) is an inherited blood disorder, where the red blood cells are abnormally shaped like a “sickle”, which is an implement with semicircular blade and a short handle. This gadget is used for cutting long grass and cereal crops. The normal red blood cell is disc shaped, and moves unhindered through the blood vessels, and lasts for 90 to 120 days before it is broken down.
However, sickle-shaped red blood cells clog together inside blood vessels, and obstruct smooth blood flow. These sickle-shaped red blood cells are also more fragile, and lasts only 10 to 20 days.1 Therefore, they are unable to adequately perform the primary function of supplying oxygen to different parts of the body.
Haemoglobin is a substance contained within the red blood cells (also called erythrocytes). It is the haemoglobin that specifically performs the function of oxygen transportation that is attributed to erythrocytes, and also gives blood it’s red colour. In most humans, offspring inherits one haemoglobin gene from each parent, making up the two, which determine the genotype of the individual.
This genotype determines whether the person will have sickle cell disease or not. There are rare types of adult haemoglobin such as Haemoglobin C, D, E, and Beta-thalassaemia haemoglobin; but the dominant ones that determine most cases of sickle cell disease are haemoglobin A, and mutant hamoglobin S. Haemoglobin A gene is normal, while haemoglobin S gene is abnormal.
An offspring inherits haemoglobin A or S from each of the parents, and this makes up the haemoglobin genotype of the individual. Hence, the combination of the genotype for individuals are AA, AS, or SS. Persons with AA genotype are normal, and do not have problems associated with the S gene. They do not carry the S gene, and thus cannot pass it to offspring.
However, persons with AS genotype are carriers of the sickle cell trait, and can pass the S gene to an offspring. Persons with SS genotype are said to have sickle cell disease, and usually inherited when the two parents have AS or SS genotype.
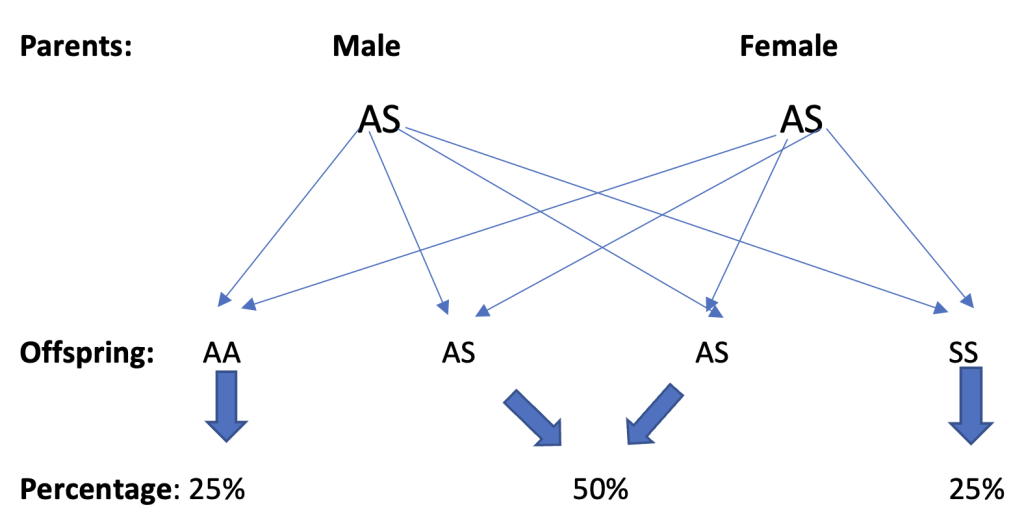
For each pregnancy, in a situation where both parents have AS genotype, there is 1 in 4 chance (25%) that the offspring will have AA genotype, 1 in 2 chance (50%) that the genotype of the offspring will be AS, and 1 in 4 chance (25%) that the offspring will have SS genotype.
So, for every pregnancy resulting from intercourse between two persons with AS genotype, there is a 25% possibility that the offspring will have sickle cell disease.2
In most cases, people who are sickle cell trait carriers do not manifest any sickle cell disease symptom. However, in rare cases symptoms and complications may manifest in these carriers when exposed to certain situations that exert high oxygen demand, such as very strenuous physical activities.
Combination of haemoglobin S with rare types haemoglobin C (SC), D (SD), E (SE), or haemoglobin beta-thalassaemia, also results in sickle cell disease.1 Below is a diagram that illustrates the probability of inheriting sickle cell disease from parents that both have AS genotype (trait carriers).
Sickle cell disease condition was first described in medical literature by American physician called James B. Herrick in1910.3 Other scientists, E. A. Beet and J. V. Neel in 1949, determined the genetic transmission of the disease; while the protective effect of Sickle cell trait against malaria was described in 1954.4
This disease is found mostly among people of African descent, though it is also found in India, Saudi Arabia, the Mediterranean, and Southern Europe.2 As high as 66% of the 120 million people living with sickle cell disease globally are in Africa, with 6.4% of under-5 mortality in Africa being attributed to SCD.
As high as 50 – 80% of infants born with SCD in Africa, die before the age of 5 years.5 Nigeria is the most sickle cell endemic country in Africa, with SCD contributing 8% of annual infant mortality in the country.
It is also estimated that about 24% Nigerian adults are sickle cell trait carriers.6 Generally, patients with SCD used to have short lifespan as seen in them, living averagely for 14 years in the early 1970s; but today they could live into their fifties and beyond.7
See Also: Breast Cancer (Symptoms, Diagnosis, Preventions, Treatment, etc)
What are the Risk factors for Sickle Cell Disease?
Incidentally, the definite risk factor for SCD is genetic, which is non-modifiable. The two parents must have AS genotype, or one will have AS, while the other will have SS; to be able to produce an offspring that has SCD. In a broader sense, race is a key non-modifiable risk factor, since the disease is commoner among people of African descent. In the United States of America, it is commoner among black Americans.7
For persons who already have SCD, tobacco smoking and excessive alcohol intake are among modifiable risk factors that could make the disease condition worse, and lead to complications. Others include;
A. For Vaso-occlusive crisis (Associated with blockage of blood vessels. It is also called pain or thrombotic crisis):8
- Extremes of temperature (heat or cold)
- Physical exertion
- Infection
- Emotional stress
- Dehydration
B. For Osteonecrosis (Associated with necrosis of the bone):2,8
- All Vaso-occlusive crises risk factors
- High body mass index
- Male sex
- Old age
C. For Neurological complications (Associated with development of cognitive impairment, spinal cord injury or stroke)
- Anaemia
- Low oxygen saturation
D. For Acute Anaemic Crisis [Hyperhaemolytic or Aplastic crisis] (Associated with rapid drop in haemoglobin level)
- Folic Acid deficiency
- Bone infarction
- Infection
- Parasitic infestation
- Medications that are toxic to bone marrow, such as Phenylbutazone
E. For Acute chest syndrome (Associated with respiratory distress)
- Asthma
- Infection
- Overexertion, resulting in hypoxaemia
What are the Signs and Symptoms of Sickle Cell Disease?
The manifestations of SCD in children usually do not present before the fifth month of life, because fetal haemoglobin is still present in large quantity; and protects the red blood cells from sickling.2 Early manifestation of SCD may include hand-and-foot syndrome, where the hands and feet are swollen, and commonly associated with pain.2
Other signs and symptoms include;1,2,9
- Recurrent pains: This usually results from sickle-shaped red blood cells blocking tiny blood vessels and impeding blood flow. This frequently happens in the bones and abdomen.
- Pallor: Anaemia is common as a result of very short life-span of the red blood cells.
- Recurrent infections, presenting as fever: SCD damages the spleen, which helps in fighting infections, thus leaving the person more vulnerable to infections. The person’s immunity is generally reduced.
- Stunted growth: Red blood cells (RBC) also transport nutrients to different parts of the body, thus enhancing growth. Reduction in the number of functional RBCs leads to a decrease in nutrients available for growth.
- Impaired vision: Tiny blood vessels in the eye, especially the retina can be blocked by sickled cells, leading to vision impairment.
- Persistent jaundice: As a result of frequent breakdown of RBCs, and the release of bilirubin which colours the white of the eye, and skin yellow; the eyes and skin frequently have a tinge of yellow discolouration.
- Severe weakness and lethargy: The body is not getting enough oxygen and nutrients, because of paucity of healthy RBCs. The body thus becomes weak.
- Enlargement of the spleen: The spleen is located at the left side of the abdomen. In addition to fighting infections, it also helps in filtering the blood, and removing dead RBCs. These RBCs could get trapped in the spleen, causing painful enlargement of the spleen.
- Dactylitis: This is inflammation and swelling of the bones of the fingers and toes, as a result of infection. This condition can also be seen in Rheumatoid arthritis, tuberculosis infection of the bones of the fingers and toes, and autoimmune diseases.
How does one make a Diagnosis of Sickle cell Disease?

Diagnosis of SCD can be made at different stages of life, including intra-uterine period, newborn, childhood and adult life. Blood and genetic tests are used in making diagnosis of SCD. Simple blood sickling test can reveal if someone has sickle cell haemoglobin, but is not definitive for SCD, since it could be positive for trait carriers.
Prenatal period: Intra-uterine diagnosis of SCD can be made using amniotic fluid sample at 14 to 16 weeks of pregnancy, or placenta specimen as early as 10 to 12 weeks.10 Prenatal testing does not reveal the potential severity of the disease.
Newborn period: Drop of blood is taken from the heel of the newborn, through needle-prick. The blood is then tested for sickle cell haemoglobin. Routine newborn screening for SCD is done in some developed countries.11
Children and adults: Blood test for genotype, commonly done through electrophoresis method is usually reliable in making diagnosis of SCD in children and adults. The blood sample is usually collected from the vein of the patient that is suspected of having SCD.10
In some high-income countries, Transcranial Doppler ultrasound screening for all children with SCD, aged two years and above, to identify children at high risk of developing primary stroke.12
How is Sickle Cell Disease Prevented?
Prevention of SCD can be categorized into primary, secondary, and tertiary levels.
Primary level: The mainstay of SCD prevention, rests on preventing persons with sickle cell trait, from making babies. Genetic counseling and pre-marital screening for intending couples, are invaluable tools in preventing SCD in any community.
Ascertaining the genotype of children, prior to entry into schools is also a good preventive measure, since the children would be aware of their genotype status early in life; and hence, empowered to start avoiding relationship with partners, who potentially could make them beget a SCD offspring.
Governments and Non-governmental organizations are encouraged to mount good health promotion programmes, that provide information to the public; on how people can primarily avoid producing children with SCD.
Secondary level prevention: Early diagnosis, and initiation of treatment contribute a lot to the prognosis of SCD. For this reason, newborn screening for SCD is advocated. Once diagnosis is made early, and proper steps are taken to prevent complications.
Tertiary level prevention: At the tertiary level of prevention, efforts are made to rehabilitate SCD patients who have developed complications. Special efforts are also made to limit the effects of disabilities arising from SCD-associated complications, and also to provide palliative care for those with bad prognosis.
What are the Complications of Sickle Cell Disease?
Sickle cell disease affects different organs and parts of the body, leading to complications that could be life-threatening. The key complications include;
Pain crisis
Also called Vaso-occlusive or thrombotic crisis, which is caused by sickled RBCs blocking the blood vessels. This is seen more on long bones, abdomen, chest, and back.2 This crisis can be triggered by temperature changes (cold or hot), illnesses, altitude, or stress.1
Anaemic crisis
This is of two types, namely Hyperhaemolytic crisis and Aplastic crisis. Hyperhaemolytic crisis is usually precipitated by infection, while Aplastic crisis is essentially as a result of failure of the haemopoietic system responsible for production of RBCs.
This failure could be due to bone marrow infarction or infection.2 Other forms of anaemic crisis are; Sequestration crisis in which the spleen is usually enlarged, Megaloblastic anaemia due to folate deficiency, and iron deficiency anaemia due to parasitic infestation or inadequate dietary intake. Iron deficiency anaemia is usually rare.2
Acute chest syndrome
Intrapulmonary sequestration of sickled RBCs could predispose the SCD persons to different types of severe chest infections.
Organs and body parts injury
Major organs and body parts affected by SCD are;
- Spleen: Injury to the spleen arises from destruction and accumulation of sickled RBCs. This leads to a condition known as Acute splenic sequestration crisis. It could be very distressful, and also lead to anaemia.1,2
- Liver and gallbladder: Liver and gallbladder are often, adversely affected in SCD patients, because of the role of the liver in breakdown of RBCs and the formation of bile. The following complications are common among SCD patients;1,2
- Cholecystitis
- Cholelithiasis
- Choledocholithiasis
- Biliary sludge
Gall stone (Commoner in adult, among whom up to 75% of SCD patients are affected)
- Lungs: The lungs of SCD patients could also be adversely affected, leading to pulmonary hypertension, and lung infections. Clotting of blood in the lungs could also lead to pulmonary embolism.
- Eye: The sickle cell RBCs can block the tiny blood vessels in the eye, causing vision impairment. The retina is most commonly affected, leading to what is called sickle retinopathy. This ultimately could lead to retinal detachment.1
- Ear: Obstruction of blood flow, and deprivation of oxygen and nutrients to any part of the ear could lead to hearing impairment. This is occasionally seen among SCD patients.
- Kidney: This is another organ where the damage caused by the obstruction to blood flow, can lead to very serious health challenge for SCD patients. This may lead to frequent kidney infections, and kidney failure.1
- Bone: The bone is also a common site of infection for SCD patients. This results in recurrent attacks of osteomyelitis if proper prophylactic measures are not taken. Salmonella bacteria organism is the most notorious organism that causes osteomyelitis in SCD patients. Impairment of blood supply to the bones, as a result blocked blood vessels could lead to avascular necrosis of the bone. This is more frequently seen in the hip bone.
- Leg: Leg ulcer are commonly seen in SCD patients that are ten years and above. These ulcers usually start small, and increase in size, if not properly managed.1
Decreased immunity
Injury to the spleen, which is part of the reticuloendothelial system that plays major in fighting infection; exposes the SCD patient to frequent infections.1
Priapism
Priapism is a known complication of SCD. This is a situation where there is prolonged and painful erection of the penis. In most cases it occurs in the absence of sexual stimulation, though it could be initiated by sexual arousal. It is classified into two types;
- Stuttering type that lasts for 4 hours or less, resolving spontaneously, and
- Major or Fulminant type, which lasts above 4 hours
Priapism can cause permanent damage to the penis, leading to erectile dysfunction.1
Cerebrovascular accident
Obstruction of smooth blood flow in the brain due to clogging of the sickled RBCs, causes stroke in many cases. Some of these may present as silent brain injuries, that gradually lead to cognitive impairment.1,2
Progressive narrowing of the brain blood vessels, prevents enough oxygen from getting to the brain. Stroke could manifest as brain infarction in children, or cerebral haemorrhage in adults.
Cardiovascular system problems
Obstruction of the blood vessels in different parts of the body could lead to problems such as hypertension and cardiac failure.2 Chronic anaemia seen frequently in SCD patients, also leads to anaemic heart failure.
Clotting of blood, as a result of sticking together of the sickle cell RBCs could also lead to deep vein thrombosis.
Delayed growth
This results from reduced ability of the RBCs to transport nutrients and oxygen to different parts of the body.
See Also: Cervical Cancer (Symptoms, Types, Diagnosis, Prevention, Treatment, etc.)
How is Sickle Cell Disease Managed?
Good management of SCD entails taking proper steps, to ensure that complications associated with the diseases are minimized or effectively managed, as they present; and the patient’s life prolonged as much as is possible. These steps include;1,2,6,13-25
Making early diagnosis and following-up the person
Once the diagnosis is made, interventions that will prevent complications are promptly initiated. If early interventions are not initiated in many SCD children, the problem could act as a swift and invisible killer within the first few years of life. The death could result from bacterial sepsis, or acute splenic sequestration crisis.
Providing information to the parents/caregivers and public, through health education and counselling
In addition to general health education and public health information on SCD, that should be provided by governments and non-governmental organizations through mass media; parents/caregivers of SCD children should be given essential information on SCD as soon the diagnosis is made, so that they are properly equipped to care for the children.
This parental/caregiver SCD health education programme, should be continued throughout the childhood of the affected child/children. The health education should be re-enforced at every clinic visit. The information given to parents/caregivers should include;
- Signs/symptoms of complications
- Prevention of complications
- Importance of regular health checkup.
- The parents/caregivers should also be taught how to palpate the spleen, so that splenic sequestration crisis can be picked up early.
Preventing infections among SCD patients
As a result of injury to the spleen, the immunity of SCD children is reduced, thus exposing them to frequent bacterial infections with Streptococcus pneumoniae and Haemophilus influenzae. The risk of these infections is more in the first five years of life.
Oral Penicillin prophylaxis given to children less than three years, protected them against these infections in 84% of cases. This oral Penicillin is also given up to five years and beyond. Oral Penicillin is given 125mg, twice daily from about three months of age. This dose should be increased to 250mg as from three years of age. Erythromycin is used in place of penicillin, if there is Penicillin allergy.
Some international programmes use intra-muscular penicillin monthly, in place of the oral one; in other to ensure compliance. Penicillin prophylaxis may be discontinued after the age of five years. The incidence of invasive Pneumococcal disease among SCD young children decreased by 93.4%, after the introduction of protein-conjugated pneumococcal vaccines.
Other vaccines recommended for SCD children are H. Influenzae type b, and Meningococcal conjugate vaccine. It is also very important that SCD children receive complete routine immunization. Sickle cell disease patients are also prone to frequent malaria attack.
It is advised that they receive Proguanil prophylaxis. The recommended dose is 50 – 100mg/day for children up to fifteen years of age, and 200mg/day for adults.
Prevention of anaemia
Sickle cell disease patients are prone to megaloblastic anaemia. It is recommended that they take 2.5 – 5mg daily dose of Folic Acid tablet to prevent anaemia.
Treating other illnesses and complications promptly, as they present
Urgent erythrocyte transfusion is also very helpful in managing most SCD complications. Acute Vaso-occlusive crisis is usually managed with appropriate analgesic drugs such as non-steroidal, or narcotic analgesics.
Nonpharmacologic interventions such as oral hydration, heat, massage, and some self-relaxation techniques may also be used in managing acute Vaso-occlusive crisis.
In addition to drugs used for relieving pain in acute Vaso-occlusive crisis, oxygen may be used in cases of Acute chest syndrome, if associated with hypoxia. Acute chest syndrome could be life-threatening, and associated with respiratory distress, fever and anaemia.
Spirometry may be indicated in Acute chest syndrome. Radiological investigation may reveal chest infection, in which case good quality, broad spectrum antibiotics is indicated. Bronchodilators may also be used in selected cases.
Acute splenic sequestration crisis lowers the immunity of the child, since the spleen belongs to the reticuloendothelial system, and involved in fighting infections. Analgesics are also used to manage this complication. Intermittent blood transfusion is also very helpful.
Surgical removal of the spleen could be the last resort, in very worrisome and recurrent episodes. Benefits of removal of the spleen should be weighed against the potential problem of recurrent infections.
Incidence of cerebrovascular complications is high among SCD persons. There is stroke incidence of about 11% by 20 years of age, and 24% by 45 years of age, among SCD adults. In cases of frank stroke, blood transfusion and moderate intravenous fluid have been found to be useful.
In certain critical situations, exchange blood transfusion could be done to reduce the level of haemoglobin S in circulation. Monthly transfusion (commonly packed cells only) helps in preventing primary and secondary stroke.
Osteomyelitis is a common bone infection that occurs in SCD persons. Detecting this infection early contributes to better management outcome. There should be high index of suspicion, if the patient complains of recurrent pain on a particular spot, especially the limbs.
The usual diagnostic processes for osteomyelitis, including X-ray should be commenced immediately. If the infection is resistant to the routine broad-spectrum antibiotics, then swab should be taken from the wound for culture and sensitivity test.
Priapism is a worrisome complication of SCD. Initial management of priapism could consist of encouraging the patient to urinate and take fluids. If it is not resolving, Etilefrine drug could be used, and the dose gradually increased. Cyproterone tablet may be added if the patient is not responding to Etilefrine. Very troublesome cases could be treated surgically.
Care of the adult SCD person
Many SCD children now survive into adulthood and some into good old age, as a result of improved care for SCD children. This has now thrown up the challenge of guidelines and practices that will further ensure that these persons keep improving on their lifespan.
It is now recommended that adult SCD persons receive immunization boosters for pneumococcal, meningococcal, and varicella vaccines. Some additional health challenges associated with adult SCD patients include difficult leg ulcers, hepatic, endocrine, and cardiac damage related to transfusion iron overload.
Investigations aimed at early detection of injuries to any of the body organs should be conducted at regular intervals. It is important to note that Blood pressure and serum creatinine of SCD adults are usually lower when compared to other adults. This implies that values at the upper limit of normal, may actually be pointing to renal dysfunction.
Care of the pregnant SCD female
In pregnancy, SCD exposes females to more challenges and dangers. The pregnant females are vulnerable to all the complications associated with the disease, and these increase their chances of having spontaneous abortion, intrauterine growth retardation, and pre-eclampsia/eclampsia. Other health challenges seen in pregnant SCD females include; venous thromboembolism, urinary tract infection and dehydration.
Care of the pregnant SCD female, ought to commence with pre-conception counselling. Early Antenatal care booking is advised, and the pregnant female could be more frequent in antenatal visits, than the general recommendation for other healthy females.
New developments in the treatment of SCD
A lot of research is currently going on, with respect to permanent cure of SCD. Bone marrow transplantation and stem cell therapy are said to have been successfully conducted is some situations. However, four medicines have been approved to treat SCD. These are;
- Hydroxyurea, which was the first to be approved in 1998. It is an oral drug that reduces sickling, and also prevents many complications. This medicine is contraindicated in pregnancy.
- Voxelotor is used in children aged four years and above, as well as adults. It also prevents sickling and binding together of RBCs.
- L-glutamine is approved for patients aged five years and above. It helps in lowering pain crisis.
- Crizanlizumab-tmca is given intravenously, and also helps in preventing sickling and pain crisis. It is used in patients aged sixteen years and above.
Healthy lifestyle modification for SCD persons
Lifestyle can be said to be, the typical way individuals or group of people live. It includes habits, diet, occupation, values, attitudes, practices, physical activities, how stressful situations are managed, relationships, sleep pattern, use of harmful substances, social and environmental situations.
It is very important that SCD patients adopt lifestyles that protect them from complications of the disease, and enable them live longer. Some of these healthy lifestyles include;
- Good quality sleep
- Drinking enough water daily
- Moderate, but not strenuous physical activities
- Avoiding stressful situations, and managing them well as they arise
- Good nutrition
- Avoidance of tobacco smoking
- Avoidance of alcohol consumption
- Avoidance of drug abuse
- Avoidance of harmful substances such as illicit drugs and Marijuana
- Deliberately staying off risky habits, behaviours and practices
- Cultivating healthy relationships and connections
- Regular health checkup visits
- It is also very important that SCD persons, avoid cold or hot environments
What are the Myths and Misconceptions about Sickle Cell Disease?
These are erroneous and misleading opinions, perceptions, and practices about SCD, that people accept. Some of these misconceptions, especially in the low- and middle-income countries are;1,26-30
- SCD is a punishment from God: This disease results purely from inheritance from both parents. It is a genetic disease that has nothing to do with God’s punishment.
- Witchcraft can be used to inflict SCD on children: Witchcraft has nothing to do with SCD. Genetic make-up of both parents determines the possibility of the offspring inheriting SCD.
- SCD patients should not socialize with people who are free from the disease: Stigmatization against people with SCD should be very strongly condemned. Socializing, and integrating fully with people is essential for wellbeing, and better quality of life for the SCD person.
- Certain environmental factors could cause SCD: No environmental factor could cause development of SCD.
- SCD affects only black skin-coloured people: It is true that this disease affects predominantly the black race, any other race can also be affected. It is found in India, Saudi Arabia, the Mediterranean, and Southern Europe.
- SCD is contagious: This disease cannot be acquired through contact with someone who has the disease.
- SCD patients are always lazy: This is not true. Although, some may be chronically weak because of reduced supply of oxygen and nutrients to the tissues, and possibly anaemia; that does not mean that they are lazy. Many are so committed to whatever endeavours they are involved in, and achieve steady results.
- People with SCD do not live into adulthood: Many people with SCD have lived way beyond adulthood, into old age. Proper care, good management of complications as they arise, and adoption of healthy lifestyle ensure that persons with SCD live long.
- SCD persons are immune to malaria: Persons with SCD are actually endangered by malaria. That is why it is recommended that they take malaria chemoprophylaxis. Persons with sickle cell trait (AS) however, have some level of immunity against malaria.
- SCD patients can never have children of their own: Pregnancy in persons with SCD could be challenging in many cases, but the outcome in most cases is good, if the pregnancy and delivery is professionally handled. SCD reduces the fertility level in both females and males, but many of them eventually have children.
- SCD patients are all intellectually impaired: Intellectual capacity of SCD persons is the same with that for persons without SCD. This is why you find among people in social class 1, such as doctors, lawyers, engineers, etc.
- Some children can outgrow SCD: One does not get remission from SCD as the person is getting older. Once the person’s genotype is SS at birth, he/she will keep producing sickle-shaped RBC, unless he/she has had successful bone marrow transport.
- Cure of SCD is achieved by use of certain herbs: Research is ongoing about developing SCD remedy from herbs, as is done with many orthodox medications; there is however no herbal concoction that is proven to cure SCD. The claims are false.
- Physical activities and exercises are prohibited for people with SCD: This is not correct. Physical activities and exercises are recommended lifestyle for SCD persons. This however, must be undertaken cautiously, so that the person is not tipped into any form of crisis
- Prayer houses can cure SCD: Prayer houses do not cure SCD. Some persons who believed they could get cure from prayer houses, ended up dying in those places.
Message from Community Positive Health Attitude Initiative
Sickle cell disease is completely preventable. All hands should be on deck, to ensure that no baby is born with SCD. The key is in mounting good public awareness campaign, and ensuring that all intending couples do their genotype test, and do not get married if both carry the sickle cell gene.
Every legal means should be used to persuade couples who potentially could produce SCD baby, not to get married. It is disturbing that some understand the risk involved and still go ahead with the marriage, asserting that “it is not their portion”, since the God they serve will not allow them to have SCD baby.
They are usually carried away by the physical pleasure they derive from each other, at that initial stage. Experience has shown that the “love” feelings start waning drastically as soon as one SCD baby arrives. Blame game then sets in.
References
- https://www.nhlbi.nih.gov/health/sickle-cell-disease (Accessed, 23/07/2024)
- Azubuike JC, Nkanginieme KEO. Paediatrics and Child Health in a Tropical Region, 3rd Edition. Educational Printing and Publishing, Lagos, Nigeria; 2016.
- Savitt TL, Goldberg MF. “Herrick’s 1910 case report of sickle cell anemia. The rest of the story”. The Journal of the American Medical Association. 1989; 261 (2): 266–271. doi:10.1001/jama.261.2.266. PMID 2642320.
- Serjeant GR. “One hundred years of sickle cell disease”. British Journal of Haematology. 2010; 151 (5): 425–429. doi:10.1111/j.1365-2141.2010.08419.x. PMID 20955412
- World Health Organization. Integrated Africa Health Observatory. Sickle Cell Disease: the silent killer in Africa. Analytical Fact Sheet. May 2024. https://files.aho.afro.who.int/afahobckpcontainer/production/files/Regional_Factsheet_on_Sickle_Cell_Disease_EN.pdf (Accessed, 23/07/2024)
- Federal Ministry of Health. National Guideline for the Control and Management of Sickle Cell Disease. Fed Min of Health, Abuja, Nigeria; 2014.
- National Heart, Lung, and Blood Institute. A Century of Progress. Sickle Cell Disease: Milestones in Research and Clinical Progress. https://www.nhlbi.nih.gov/sites/default/files/publications/Sickle_Cell_Milestones_in_Research_508_Updated_0.pdf (Accessed, 23/07/2024)
- https://www.rarediseaseadvisor.com/hcp-resource/sickle-cell-disease-risk-factors/ (Accessed, 23/07/2024)
- Litin S. Mayo Clinic Family Health Book, 5th Edition. Mayo Clinic Press, United States of America, 2018.
- https://www.msdmanuals.com/professional/hematology-and-oncology/anemias-caused-by-hemolysis/sickle-cell-disease (Accessed, 23/07/2024)
- https://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia/diagnosis-treatment/drc-20355882 (Accessed, 23/07/2024)
- Adams R, McKie V, Nichols F, Carl E, Zhang DL, McKie K, Figueroa R, Litaker M, Thompson W, Hess D. 1992. The use of transcranial ultrasoundography to predict stroke in sickle cell disease. N Engl J Med 326: 605–610.
- Pearson HA. Sickle cell anemia and severe infections due to encapsulated bacteria. J Infect Dis, 1977; 136: S25–S30.
- Pearson HA, Spencer RP, Cornelius EA. Functional asplenia in sickle-cell anemia. N Engl J Med, 1969; 281: 923–926.
- Powars D, Overturf G, Weiss J, Lee S, Chan L. Pneumococcal septicemia in children with sickle cell anemia. Changing trend of survival. JAMA, 1981; 245: 1839–1842.
- Rogers DW, Clarke JM, Cupidore L, Ramlal AM, Sparke BR, Serjeant GR. Early deaths in Jamaican children with sickle cell disease. Br Med J, 1978; 1: 1515–1516.
- Winkelstein JA, Drachman RH. Deficiency of pneumococcal serum opsonizing activity in sickle cell disease. N Engl J Med, 1968; 279: 459–466.
- Gaston MH, Verter JI,Woods G, Pegelow C, Kelleher J, Presbury G, Zarkowsky H, Vichinsky E, Iyer R, Lobel JS, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. N Engl J Med, 1986; 314: 1593–1599.
- Halasa NB, Shankar SM, Talbot TR, Arbogast PG, Mitchel EF, Wang WC, Schaffner W, Craig AS, Griffin MR. Incidence of invasive pneumococcal disease among individuals with sickle cell disease before and after the introduction of the pneumococcal conjugate vaccine. Clin Infect Dis, 2007; 44: 1428–1433.
- McGann PT, Nero AC, Ware RE. Current Management of Sickle Cell Anemia. Cold Spring Harb Perspect Med, 2013; 3: a011817
- Rees DC, Olujohungbe AD, Parker NE, Stephens AD, Telfer P, Wright J. 2003. Guidelines for the management of the acute painful crisis in sickle cell disease. Br J Hematol, 2003; 120: 744–752.
- Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S,Moohr JW,Wethers DL, Pegelow CH, Gill FM. Cerebrovascular accidents in sickle cell disease: Rates and risk factors. Blood, 1998; 91: 288–294.
- Swerdlow PS. Red cell exchange in sickle cell disease. Am Soc Hematol Educ Program, 2006; 48–53.
- https://en.wikipedia.org/wiki/Sickle_cell_disease (Accessed, 23/07/2024)
- Ndibuagu EO. Long, Healthy and Happy Living: Key Lifestyle Paths. NEDAL Publishers, Enugu, Nigeria; 2024
- https://www.sicklecellspeaks.com/managing-sickle-cell/overcoming-misconceptions (Accessed, 23/09/2024)
- https://sickle-cell.com/myths (Accessed, 23/09/2024)
- https://sicklecellanemianews.com/columns/myths-facts-about-sickle-cell-disease-stigma-discrimination/ (Accessed, 23/09/2024)
- https://bastionhmo.com/sickle-cell-disease-facts-and-myths/ (Accessed, 23/09/2024)
- https://www.surjen.com/blog-details/understanding-sickle-cell-disease (Accessed, 23/09/2024)